Welcome to STOGQY's Blog!
-Hey--
SHELL学习
基本用法
- {} () [] [[]]
{}跟python的差不多,${var}等价于$var。 ()在脚本中等价于反引号,可增加代码的可读性。 方括号语句为[ “$HOME” == “$pwd” ],注意里面的空格。在bash里面[]里面用== > 之类的似乎不用转义,但在zsh里面须用/转义。 此时用[[]]就可以不用转义直接使用 == < >等比较符。所以一般用[[]]多。
- if语句
if [条件]; then
fi - while while [conditions] do done - for for var in con1 con2 con3 ... do done for ( ( i=1; i<=$var; i=i+1 ) ) do done - case case $var in "one" ) ;; "two" ) ;; "three" ) ;; esac ### 引号 单引号不转义,里头改啥就是啥 双引号转义,可以装变量进去 反引号则执行局部代码 ### 关于参数 $0 脚本本身 $1 第一个参数 $n 第n个参数 $# 参数的个数 $@ "$1" "$2" ... $* "$1 $2 $3 ..." shift 参数往后滑移一位
-
MySQL学习
基本用法
登录
mysql -u <user name> -p -h <host name>如过没设置密码就跳过-p,如过本地登录就省掉<host name>。 root用户默认是没有密码的,通过该命令可设置密码:mysqladmin -u root password "your_new_password"root用户登录后可创建新用户:CREATE USER 'username'@'localhost' IDENTIFIED BY 'your_password';创建数据库
MySQL的语法其实就是平铺直叙的文本,通常用大写表示命令。每个独立命令须用
;表示结尾,可以随意换行,跟C语言一样。 显示已有数据库:SHOW DATABASES;使用某数据库:USE <database name>创建数据库:CREATE DATABASE <database name>( // <col name> <data type> <constrain> id varchar(60) primary key, seq text )删库:DROP DATABASE <database name>操作表
#查看表中前几行: SELECT * FROM <table name> LIMIT <row count> #查看表信息: DESCRIBE <table name> #查询: SELECT * FROM <table name> WHERE <condition> #删除表: DROP TABLE <table name> #删除数据: DELETE FROM TABLE WHERE <condition> #清空 TRUNCATE <table name> #或者 DELETE FROM <table name> # 前者快速删除不可回滚,后者逐条删除,可回滚。 # 查看表信息 SHOW TABLE STATUS LIKE <table name> \G约束
用于限制表中的数据,为了保证表中数据的准确性和可靠性,不符合约束的数据,插入时就会失败。
名称 作用 NOT NULL 非空,用于保证该字段的值不能为空 DEFAULT 默认值,用于保证该字段有默认值 PRIMARY KEY 主键,用于保证该字段的值具有唯一性并且非空 UNIQUE 唯一,用于保证该字段的值具有唯一性,可以为空 CHECK 检查约束(MySql不支持),检查字段的值是否为指定的值 FOREIGN KEY 外键,用于限制两个表的关系,用于保证该字段的值必须来自于主表的关联列的值,在从表添加外键约束,用于引用主表中某些的值 导入数据
# 逐行导入数据 INSERT INTO <table name> (<col1>, <col2>, <col3> ... ) VALUES (<value1>, <value2>, <value3> ...) # 更新数据 UPDATE <table name> SET `<col name>` = <value> `<col name>` = <value> where <condition> #如过不设置codition就对整张表进行更新 # 删除数据 DELETE FROM TABLE WHERE <condition> # 批量导入,确保local-infile 设置为on set global local_infile=on # 或者在登录的时候加上 --local_infile=1 # 查询local-infile 状态 SHOW GLOBAL VARIABLES LIKE "local_infile" # 然后就可以导入了 LOAD DATA INFILE <file path> INTO <table name> FIELDS TERMINATED BY ',' LINES TERMINATED BY '\n' ENCLOSED BY '"' CHARACTER SET utf8 IGNORE <num> ROWS # 使用source file.sql批量导入 # 设置关键参数 set sql_log_bin=OFF;//关闭日志 set autocommit=0;//关闭autocommit自动提交模式 START TRANSACTION;//开启事务 source <path to .sql file>参考信息源
-
多单拷贝基因串联构建物种发育树
本文参考自https://www.jianshu.com/p/25e60508a08f, 原作者为:生信小白2018
基本流程
单拷贝同源基因寻找 → 单基因多序列比对 → 串联后构建发育树
所需软件
- Orthofinder: 寻找单拷贝基因
- MAFFT: 多序列比对
- TRIMAL: 多序列比对修剪
- TRIMAL or FastTree: 串联法(concatenation)构建蛋白质系统发育树
- ASTRAL: 联合/合并法(coalescence)构建系统发育
流程解析
通过Orthofinder寻找单拷贝同源基因
以下内容参考自改文章:「基因组学」使用OrthoFinder进行直系同源基因分析
OrthoFinder的分析过程分为如下几步:
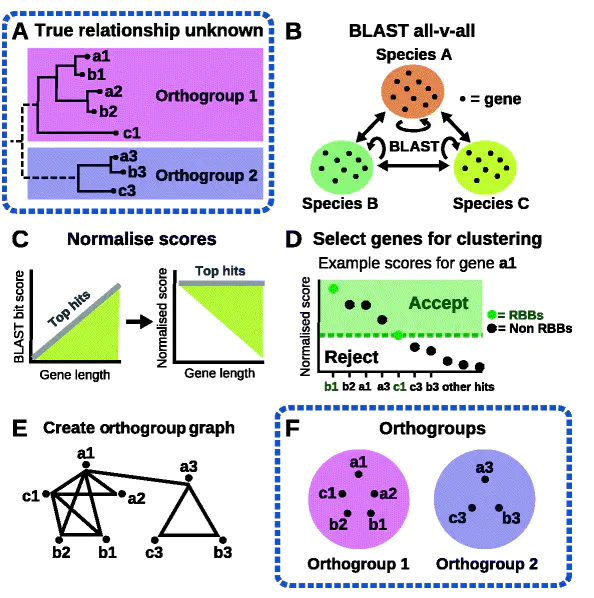
- BLAST all-vs-all搜索。使用BLASTP以evalue=10e-3进行搜索,寻找潜在的同源基因。(除了BLAST, 还可以选择DIAMOND和MMSeq2)
- 基于基因长度和系统发育距离对BLAST bit得分进行标准化。
- 使用RBNHs确定同源组序列性相似度的阈值
- 构建直系同源组图(orthogroup graph),用作MCL的输入
- 使用MCL对基因进行聚类,划分直系同源组
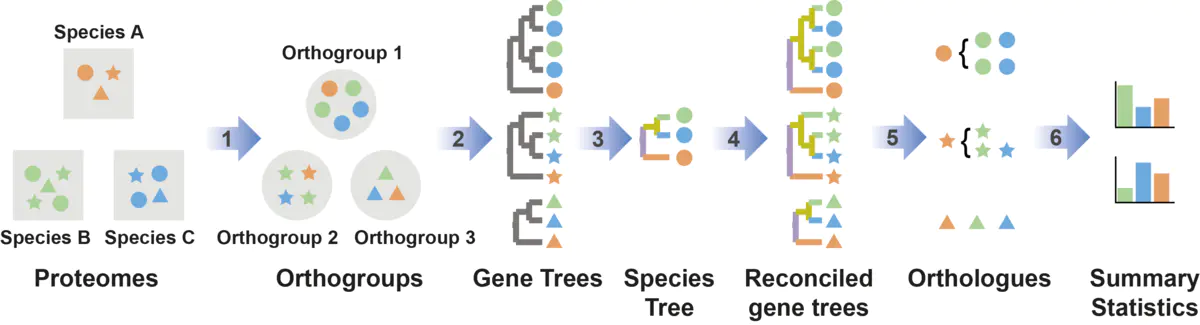
OrthoFinder2在OrthoFinder的基础上增加了物种系统发育树的构建,流程如下:
- 为每个直系同源组构建基因系统发育树
- 使用STAG算法从无根基因树上构建无根物种树
- 使用STRIDE算法构建有根物种树
- 有根物种树进一步辅助构建有根基因树
基于Duplication-Loss-Coalescent 模型,有根基因树可以用来推断物种形成和基因复制事件,最后记录在统计信息中。
OrthoFinder的使用非常方便,一行命令即可,但跑起来比较花时间:
orthofinder -f <folder contains target genomes> -S diamondOrthoFinder结果:
运行结束后,会在
ExampleData里多出一个文件夹,Results_Feb14, 其中Feb14是我运行的日期直系同源组相关结果文件,将不同的直系同源基因进行分组。
Orthogroups.csv:用制表符分隔的文件,每一行是直系同源基因组对应的基因。
Orthogroups.txt: 类似于Orthogroups.csv,只不过是OrhtoMCL的输出格式
Orthogroups_UnassignedGenes.csv: 格式同Orthogroups.csv,只不过是物种特异性的基因
Orthogroups.GeneCount.csv:格式同Orthogroups.csv, 只不过不再是基因名信息,而是以基因数。
直系同源相关文件,分析每个直系同源基因组里的直系同源基因之间关系,结果会在
Orthologues_Feb14文件夹下,其中Feb14是日期Gene_Trees: 每个直系同源基因基因组里的基因树
Recon_Gene_Trees:使用OrthoFinder duplication-loss coalescent 模型进行发育树推断
Potential_Rooted_Species_Trees: 可能的有根物种树
SpeciesTree_rooted.txt: 从所有包含STAG支持的直系同源组推断的STAG物种树
SpeciesTree_rooted_node_labels.txt: 同上,只不过多了一个标签信息,用于解释基因重复数据。
比较基因组学的相关结果文件:
Orthogroups_SpeciesOverlaps.csv: 不同物种间的同源基因的交集
SingleCopyOrthogroups.txt: 单基因拷贝组的编号
Statistics_Overall.csv:总体统计信息
Statistics_PerSpecies.csv:分物种统计信息
STAG是一种从所有基因推测物种树的算法,不同于使用单拷贝的直系同源基因进行进化树构建。
一些重要概念:
-
Species-specific orthogroup: 一个仅来源于一个物种的直系同源组
-
Single-copy orthogroup: 在直系同源组中,每个物种里面只有一个基因。我们会用单拷贝直系同源组里的基因推断物种树以及其他数据分析。
-
Unassigned gene: 无法和其他基因进行聚类的基因。
-
G50和O50,指的是当你直系同源组按照基因数从大到小进行排列,然后累加,当加入某个组后,累计基因数大于50%的总基因数,那么所需要的直系同源组的数目就是O50,该组的基因树就是G50.
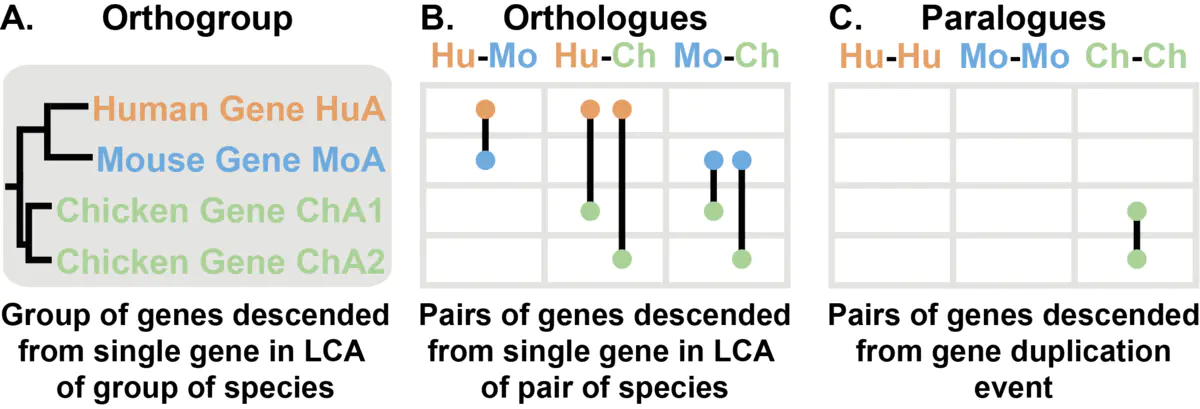
Orthogroups, Orthologs 和 Paralogs 这三个概念推荐看图理解。
- 利用MAFFT进行单同源基因的多序列比对
mafft<input.faa> > <outpuit_aligned.mafft>- TRIMAL修剪多序列比对结果
trimal -in <input_aligned.mafft> -out <input_aligned.mafft.trimed> -automated1- Concatenation法进行系统发育分析
将上述trim好的多序列比对结果按照物种顺序进行串联,然后用RaxML或者FastTree进行分析。
raxml -T <thread using> -f a -N <boostrap such as 100> -m <model such as JTT> -x 123456 -p 123456 -s <concatenated_alignment> -n <output.nwk>FastTree <concatenated_alignment> > <ouput.nwk>- Coalescence法进行系统发育分析
先用RaxML或者FastTree对每个单拷贝基因进行分析,然后用ASTRAL聚合:
# single gene tree raxml -T <thread using> -f a -N <boostrap such as 100> -m <model such as JTT> -x 123456 -p 123456 -s <SingleGene_alignment> -n <SingleGene_output.nwk> # concatenation cat <all SingleGene_alignment> >> <allSingleGenes_tree.nwk -b> # bootstrap ana cat <all bootstrap file> >> <allSingleGenes_bootstrap.txt> # ASTRAL java -jar ASTRAL -i allSingleGenes_tree.nwk -b allSingleGenes_bootstrap.txt -r <boostrap> -o <ASTRAL_out.res> > tail -n 1 <ASTRAL_out.res> > <ASTRAL_out.res.nwk>集成脚本
该脚本参考自https://github.com/dongwei1220/EasySpeciesTree, 但做了一些适当的修改
-
Mash: 使用MinHash快速估算基因距离
本文来自https://www.jianshu.com/p/cd81971b0c03, 原作者为:lakeseafly
Mash: 使用MinHash快速估算基因距离
Mash扩展了MinHash降维技术,使其成对的突变距离和P值显着性检验,从而可以有效地聚类和搜索大量序列集合。混搭将大序列和序列集还原为较小的代表性草图,进而可以快速估计基因组之前全局突变距离。MASH能用于聚类NCBI RefSeq中所有的基因组,基于组装或者它的reads的实时的基因组聚类,聚类大量的宏基因组数据集。 该工具最终发表在Genome Biology上。
软件安装与示例
#下载 wget https://github.com/marbl/Mash/releases/download/v2.2/mash-Linux64-v2.3.tar tar -xvf mash-Linux64-v2.3.tar #即可使用 #具体参数见帮助 Mash version 2.2 Type 'mash --license' for license and copyright information. Usage: mash <command> [options] [arguments ...] Commands: bounds Print a table of Mash error bounds. dist Estimate the distance of query sequences to references. info Display information about sketch files. paste Create a single sketch file from multiple sketch files. screen Determine whether query sequences are within a larger mixture of sequences. sketch Create sketches (reduced representations for fast operations). triangle Estimate a lower-triangular distance matrix.Mash 提供两个基本功能用于序列之间的比对 sketch 和 dist。 sketch 功能将序列转化为哈希结构图,生成一个.mash文件。 dist 功能比对 序列之前的sketches结果并且返回 Jaccard index的估计值P值,还有Mash距离, 这些值能用于估计一个简单进化模型的序列突变率。 因此快速比对时可以采用先生成sketch文件,然后再计算距离的方式,分步运行。
估计俩基因组的mash distance
实际上已经有更好的实现方式了,原作者在Mash的的基础上开发了FastANI,用于快速比对基因组相似性,相关研究于2018年发表在Nature communications上。
wget https://gembox.cbcb.umd.edu/mash/genome1.fna wget https://gembox.cbcb.umd.edu/mash/genome2.fna #如果一次运行: mash dist genome1.fna genome2.fna #它将返回如下结果: ###Reference-ID, Query-ID, Mash-distance, P-value, and Matching-hashes genome1.fna genome2.fna 0.0222766 0 456/1000 #还可以分步运行: mash sketch genome1.fna; mash sketch genome2.fna; mash dist genome1.fna.msh genome2.fna.msh #这样得到的结果是一样的除了基因组序列外,该功能还能直接应用到序列的reads中(fastq文件)。通过计算不同fastq文件之间的遗传距离,并将遗传距离结果整合可视化,你可以快速的得到你的不同个体测序序列之间的遗传关系: 关于如何具体建树我这里就不细说,但是给大家推荐一个非常实用的Github网站,这个网站已经将Mash构树的过程封装好为mashtree,使用起来非常方便。Github链接:https://github.com/lskatz/mashtree 不过这玩意要装的东西有点多,我没功夫弄它……
构建本地序列的mash数据库进行遗传距离分析
除了上面介绍的例子之外,mash还支持使用已知的序列构建本地的数据库,然后利用该构建的数据库,来对其它序列进行遗传距离的分析。使用刚刚下载好的genome1.fna和genome2.fna进行本地mash数据库的构建:
mash sketch -o reference genome1.fna genome2.fna构建好后可以使用info功能对构建好的数据库进行检查:
mash info reference.msh ###输出数据库的信息,证明数据库构成功: Header: Hash function (seed): MurmurHash3_x64_128 (42) K-mer size: 21 (64-bit hashes) Alphabet: ACGT (canonical) Target min-hashes per sketch: 1000 Sketches: 2 Sketches: [Hashes] [Length] [ID] [Comment] 1000 4639675 genome1.fna gi|49175990|ref|NC_000913.2| Escherichia coli str. K-12 substr. MG1655, complete genome 1000 5498450 genome2.fna gi|47118301|dbj|BA000007.2| Escherichia coli O157:H7 str. Sakai DNA, complete genome #下载新的序列,与构建好的mash数据库进行遗传距离比对: wget https://gembox.cbcb.umd.edu/mash/genome3.fna mash dist reference.msh genome3.fna ###结果显示genome3.fna与数据库的比对结果,可以发现genome1和genome3遗传具体是完全一致的(同一个样本): genome1.fna genome3.fna 0 0 1000/1000 genome2.fna genome3.fna 0.0222766 0 456/1000与已构建好的RefSeq sketch数据库进行遗传距离比对
除了自己构建mash sketch数据库外,我们当然可以下载一些已经构建好的数据库。最常用的要数RefSeq sketch数据库。
wget https://gembox.cbcb.umd.edu/mash/refseq.genomes.k21s1000.msh #由于mash没有给出测试文件,这里从我自己服务器中大豆的数据中抽取一部分进行测试: head -n 1600 SRR7817178_1.fq > test_1.fq head -n 1600 SRR7817178_2.fq > test_2.fq cat test_1.fq test_2.fq >reads.fastq #这里使用-m 2参数来进一步提高结果的质量,这个参数会忽略掉低质量的单拷贝的k-mer: mash sketch -m 2 reads.fastq #使用已下载好的RefSeq数据库,对序列的reads进行遗传相关系查询: mash dist refseq.genomes.k21s1000.msh reads.fastq.msh > distances.tab #查看结果: GCF_000004515.4_Glycine_max_v2.0_genomic.fna.gz reads.fastq 0.295981 2.36893e-05 1/1000 GCF_000297375.1_ASM29737v1_genomic.fna.gz reads.fastq 0.295981 2.25234e-05 1/1000 GCF_000400935.1_ASM40093v1_genomic.fna.gz reads.fastq 0.295981 2.16808e-05 1/1000 GCF_000400955.1_ASM40095v1_genomic.fna.gz reads.fastq 0.295981 2.16542e-05 1/1000 GCF_000837185.1_ViralProj14097_genomic.fna.gz reads.fastq 0.295981 1.63576e-05 1/1000 GCF_001411555.1_wgs.5d_genomic.fna.gz reads.fastq 0.295981 2.36883e-05 1/1000 GCF_001578535.1_ASM157853v1_genomic.fna.gz reads.fastq 0.295981 1.98188e-05 1/1000 GCF_001662445.1_ASM166244v1_genomic.fna.gz reads.fastq 0.295981 2.31897e-05 1/1000首先可以看到这些reads是和大豆(G.max)有较近的遗传距离,另外reads里面还含有一些污染,与其它细菌病毒有关系。
这里我们可以发现,mash是可以将reads的污染检测出来。事实上,mash提供了screen功能专门用于污染检测:
mash screen -w -p 4 refseq.genomes.k21s1000.msh|sort -gr |head #结果如下 0.799067 9/1000 1 6.84538e-42 GCF_001343725.1_ViralMultiSegProj188731_genomic.fna.gz [2 seqs] NC_020234.1 Rosellinia necatrix partitivirus 2 RdRp gene for RNA-dependent RNA polymerase, co mplete cds [...] 0.798763 6/672 1 2.34994e-28 GCF_000899295.1_ViralProj176433_genomic.fna.gz NC_018671.1 Sauropus leaf curl disease associated DNA beta, complete genome 0.783786 6/1000 3 2.57053e-27 GCF_001401365.1_ViralProj299179_genomic.fna.gz NC_028095.1 Torulaspora delbrueckii dsRNA Mbarr-1 killer virus strain EX1180 Kbarr-1 killer toxin gene, comple te cds 0.758374 2/666 1 2.73245e-09 GCF_000922435.1_ViralProj259986_genomic.fna.gz NC_024777.1 Small begomovirus-associated satellite isolate Sa19-S1, complete sequence 0.758338 3/1000 1 2.27756e-13 GCF_000911175.1_ViralMultiSegProj225924_genomic.fna.gz [2 seqs] NC_022616.1 Red clover cryptic virus 1 isolate IPP_Nemaro segment 1 RNA-dependent RNA polymer ase gene, complete cds [...] 0.743837 2/1000 27 6.16326e-09 GCF_001590135.1_ViralProj314638_genomic.fna.gz NC_029628.1 Lake Sarah-associated circular virus-21 isolate LSaCV-21-LSMU-2013, complete sequence 0.743837 2/1000 2 6.16326e-09 GCF_000928835.1_ViralProj268860_genomic.fna.gz NC_025790.1 Black sea bass polyomavirus 1 isolate 2835, complete genome 0.743837 2/1000 2 6.16326e-09 GCF_000888115.1_ViralProj45931_genomic.fna.gz NC_013801.1 Croton yellow vein mosaic alphasatellite, complete genome 0.743837 2/1000 2 6.16326e-09 GCF_000848385.1_ViralProj14678_genomic.fna.gz NC_001782.1 Saccharomyces cerevisiae killer virus M1, complete genome 0.743837 2/1000 2 6.16326e-09 GCF_000004515.4_Glycine_max_v2.0_genomic.fna.gz [1191 seqs] NC_016088.2 Glycine max cultivar Williams 82 chromosome 1, Glycine_max_v2.0, whole genome shotgun可以看到相对上面的结果,screen功能能够输出更加清晰的污染检测结果,包括了identity, shared-hashes, median-multiplicity, p-value, query-ID, query-comment等一系列的信息,更便于污染检测。
-
树莓派
本文持续更新
搭建aria2下载
我采用的是aria2+nginx的组合,可实现局域网内树莓派离机下载。
#下载aria2和nginx sudo apt install aria2 nginx #配置aria2 cd ~ mkdir .aria2 cd .aria2 touch aria2.conf aria2.session以下为aria2.conf的内容:
## 文件保存相关 ## # 文件保存目录 dir=/home/pi/Downloads # 启用磁盘缓存, 0为禁用缓存, 需1.16以上版本, 默认:16M disk-cache=32M # 断点续传 continue=true # 文件预分配方式, 能有效降低磁盘碎片, 默认:prealloc # 预分配所需时间: none < falloc ? trunc < prealloc # falloc和trunc则需要文件系统和内核支持 # NTFS建议使用falloc, EXT3/4建议trunc, MAC 下需要注释此项 file-allocation=none ## 下载连接相关 ## # 最大同时下载任务数, 运行时可修改, 默认:5 max-concurrent-downloads=5 # 同一服务器连接数, 添加时可指定, 默认:1 max-connection-per-server=15 # 整体下载速度限制, 运行时可修改, 默认:0(不限制) #max-overall-download-limit=0 # 单个任务下载速度限制, 默认:0(不限制) #max-download-limit=0 # 整体上传速度限制, 运行时可修改, 默认:0(不限制) #max-overall-upload-limit=0 # 单个任务上传速度限制, 默认:0(不限制) #max-upload-limit=0 # 禁用IPv6, 默认:false disable-ipv6=true # 最小文件分片大小, 添加时可指定, 取值范围1M -1024M, 默认:20M # 假定size=10M, 文件为20MiB 则使用两个来源下载; 文件为15MiB 则使用一个来源下载 min-split-size=10M # 单个任务最大线程数, 添加时可指定, 默认:5 split=10 ## 进度保存相关 ## # 从会话文件中读取下载任务 input-file=/home/pi/.aria2/aria2.session # 在Aria2退出时保存错误的、未完成的下载任务到会话文件 save-session=/home/.aria2/aria2.session # 定时保存会话, 0为退出时才保存, 需1.16.1以上版本, 默认:0 save-session-interval=60 ## RPC相关设置 ## # 启用RPC, 默认:false enable-rpc=true # 允许所有来源, 默认:false rpc-allow-origin-all=true # 允许外部访问, 默认:false rpc-listen-all=true # RPC端口, 仅当默认端口被占用时修改 rpc-listen-port=6800 # 设置的RPC授权令牌, v1.18.4新增功能, 取代 --rpc-user 和 --rpc-passwd 选项 rpc-secret=123456 ## BT/PT下载相关 ## # 当下载的是一个种子(以.torrent结尾)时, 自动开始BT任务, 默认:true #follow-torrent=true # 客户端伪装, PT需要 peer-id-prefix=-TR2770- user-agent=Transmission/2.77 # 强制保存会话, 即使任务已经完成, 默认:false # 较新的版本开启后会在任务完成后依然保留.aria2文件 #force-save=false # 继续之前的BT任务时, 无需再次校验, 默认:false bt-seed-unverified=true # 保存磁力链接元数据为种子文件(.torrent文件), 默认:false bt-save-metadata=true启动aria2:
sudo aria2c --conf-path=/home/pi/.aria2/aria2.conf -D#可将其加入自动启动 sudo update-rc.d aria2c defaults配置nginx,以下为
/etc/nginx/nginx.conf的内容:user www-data; worker_processes auto; pid /run/nginx.pid; # include /etc/nginx/modules-enabled/*.conf; events { worker_connections 768; # multi_accept on; } http { ## # Basic Settings ## sendfile on; tcp_nopush on; tcp_nodelay on; keepalive_timeout 65; types_hash_max_size 2048; # server_tokens off; # server_names_hash_bucket_size 64; # server_name_in_redirect off; include /etc/nginx/mime.types; default_type application/octet-stream; ## # SSL Settings ## ssl_protocols TLSv1 TLSv1.1 TLSv1.2; # Dropping SSLv3, ref: POODLE ssl_prefer_server_ciphers on; ## # Logging Settings ## access_log /var/log/nginx/access.log; error_log /var/log/nginx/error.log; ## # Gzip Settings ## gzip on; # gzip_vary on; # gzip_proxied any; # gzip_comp_level 6; # gzip_buffers 16 8k; # gzip_http_version 1.1; # gzip_types text/plain text/css application/json application/javascript text/xml application/xml application/xml+rss text/javascript; server { listen 80; server_name 127.0.0.1 192.168.2.76; location /aria2 { alias /var/www/webui-aria2/docs; index index.html charset utf-8,gbk; } } ## # Virtual Host Configs ## # include /etc/nginx/conf.d/*.conf; # include /etc/nginx/sites-enabled/*; } #mail { # # See sample authentication script at: # # http://wiki.nginx.org/ImapAuthenticateWithApachePhpScript # # # auth_http localhost/auth.php; # # pop3_capabilities "TOP" "USER"; # # imap_capabilities "IMAP4rev1" "UIDPLUS"; # # server { # listen localhost:110; # protocol pop3; # proxy on; # } # # server { # listen localhost:143; # protocol imap; # proxy on; # } #}启动nginx:
sudo /etc/init.d/nginx start然后在局域网内用打开
https://192.168.2.76/aria2即可,需注意在webUI里面配置好RPC令牌,即前面aria2.conf里面的rpc-secret=123456,当然可以把它改成自己的密码。
-
病原菌介导植物内生菌群抑病功能的激活
- 病原菌激活植物内生菌群的抑病功能
- 个人感想
- 内容简介
- 详细内容
病原菌激活植物内生菌群的抑病功能
Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome Science [IF:41.037]
2019-11-1 Articles
DOI: https://doi.org/10.1126/science.aaw9285
第一作者:Víctor J. Carrión1,2,Juan Perez-Jaramillo1,3, Viviane Cordovez1,2, Vittorio Tracanna4
通讯作者:Marnix H. Medema4† , Jos M. Raaijmakers 1,2†; Email: j.raaijmakers@nioo.knaw.nl (J.M.R.); marnix.medema@wur.nl (M.H.M.)
其它作者:Mattias de Hollander1, Daniel Ruiz-Buck1 Lucas W. Mendes5, Wilfred F.J. van Ijcken6, Ruth Gomez-Exposito7, Somayah S. Elsayed2, Prarthana Mohanraju7, Adini Arifah7, John van der Oost7, Joseph N. Paulson8, Rodrigo Mendes9, Gilles P. van Wezel1,2
作者单位:
1 荷兰,瓦赫宁根生态研究所,微生物生态中心(Department of Microbial Ecology, Netherlands Institute of Ecology (NIOO-KNAW), Droevendaalsesteeg 10, 6708 PB Wageningen, Netherlands)
2 荷兰莱顿大学,生物研究所(Institute of Biology, Leiden University, Sylviusweg 72, 2333 BE Leiden, Netherlands)
3 哥伦比亚共和国,安蒂奥基亚大学(PECET, University of Antioquia, Medellín, Antioquia 050010, Colombia)
4 荷兰,瓦赫宁根大学,生信中心(Bioinformatics Group, Wageningen University, Droevendaalsesteeg 1, 6708 PB Wageningen, Netherlands)
5 巴西,圣保罗大学(Cell and Molecular Biology Laboratory, Center for Nuclear Energy in Agriculture (CENA), University of Sao Paulo (USP), Piracicaba, Brazil)
6 荷兰,鹿特丹伊拉斯姆斯大学(Erasmus MC, University Medical Center Rotterdam, Department of Cell Biology, Center for Biomics, 3025 CN Rotterdam, Netherlands)
个人感想
抑病型土壤的研究主要集中在特异性抑病土壤上,目前已经有很多高水平文章解析过抑病型微生物群落,这篇Science无疑是最新和最全的一篇。从内容上来,描述了抑病型土壤的群落结构和功能;得到了候选功能菌株,并通过培养组得到了大量的分离株,并开展菌株功能验证;而后进行功能基因的筛选,敲除和验证,最终形成了一个完美的故事。确实是本领域研究的学习模板,并值得我们重现这篇文章的分析过程。
这篇文章关注的是抑病型微生物群落的解析,对于抑病型微生物群落的形成也十分重要,那么内生菌群是如何被植物招募并进入根系内部的呢?是否根系分泌物在这个过程中也起着重要作用?相信这将是很有意思的故事。
内容简介
一些土壤显示出显著的抑制植物病原体引起疾病的能力,这种能力归因于与植物相关的微生物群。 Carrión等研究枯萎真菌茄枯萎病菌感染甜菜,内生菌(在根中发现的亲密微生物群落)在真菌疾病抑制中的作用。转录分析表明,几种细菌内生菌种会激活生物合成基因簇,从而抑制疾病。这些生物产生抗真菌效应物,包括可消化真菌细胞壁的酶,以及次生代谢产物,包括吩嗪,聚酮化合物和铁载体,可能有助于抗真菌表型。
生活在植物内部的微生物可以促进植物的生长和健康,但是它们的基因组和功能多样性仍然难以捉摸。在这里,宏基因组学和网络推论表明,植物根部的真菌感染在根内富集了几丁质菊科和黄杆菌科,以及几丁质酶基因和编码未知核糖体肽合成酶(nonribosomal peptide synthetases,NRPSs)和聚酮化合物合酶(polyketide synthases,PKSs)的各种未知的生物合成基因簇。菌株级别合成的几丁质和黄杆菌的菌群,以持续抑制真菌根部疾病。然后定点诱变表明,以前未鉴定的黄杆菌中的NRPS-PKS基因簇对于内生菌群抑制疾病至关重要。我们的研究结果表明,内生根微生物群具有许多尚不为人所知的功能性状,可以共同保护植物内外。
植物微生物组研究已经积累了大量的测序数据和丰富的信息,表明了许多植物根际、叶际、种子和胚层中不同微生物群落的多样性和丰富度。然而,迄今为止,很少有研究论证微生物菌群对特定植物表型(即植物生长、发育和健康)的影响。因此,在分子和化学层面上许多植物表型与微生物群结构和功能之间在的因果关系仍然未知。本研究旨在探讨植物内生微生物菌群的基因多样性及对真菌感染植株的保护作用。为此,我们整合了包括网络推断和宏基因组学在内的多种方法,以寻找可以抑制Rhizoctonia solani枯萎病(水稻、小麦和甜菜等几种植物根部的真菌病原菌)的内生菌菌群体和功能基因簇。
抑病土壤是一种特殊的生态系统,在这里,由于微生物群落的保护作用,植物才得以存活。各种类型的抑制性土壤都相继报道过,包括真菌、细菌、卵菌和线虫的抑病土壤。抑病性可以通过加热消失,也可以移植到没有抑病作用的土壤中,类似于人类的粪菌移植。
在田间土壤中,通过感病作物的连续栽培过程中集中爆发病害来诱导土壤对病原菌(例如:R.solani)的特异性抑制作用(类似于人类的疫苗)。一旦抑病性形成,如果种植非寄主植物时,这种抑制不表现;但在寄主植物和特定真菌病原体的存在下,抑病作用重新唤醒。这种特异性抑病性土壤一旦形成,便保护之后种植的同种作物和对病原菌产生抵抗性,但是抑病性却在种植其他作物时不表现。
因此,病原菌、寄主植物及其根际微生物群之间的相互作用是特异性抑病性产生和持续的关键因素。我们以前的研究表明,对病原菌 R.solani 有抑制作用的土壤中,发现甜菜根际的几个细菌属paraurkholderia、假单胞菌(pseudomonas)和链霉菌(streptomyces)起着重要的作用。
为了了解那些生活在植物根组织中的微生物(内生菌)在抑病性产生过程中发挥的作用,我们对生长在抑病土壤中的甜菜幼苗的根内进行了宏基因组测序分析;并鉴定了与抑病相关的微生物群落和功能组成;以区分哪些生物合成基因簇(BGCs)在感染过程中上调,然后重组内生菌群;最后进行位点定向突变,检测特异性BGCs是否在抑病过程中起着重要作用。
详细内容
内生菌群落多样性和网络分析
Taxonomic diversity and network inference of the endophytic microbiome
附图1展示了研究设计:设置甜菜种植在发病(Conducive, C)和抑病土(Suppressive, S)壤和是否接种病原菌R. solani(R)共计四个处理。在抑病土壤中接种病原菌(S+R)的甜菜发病率为15-30%,在感病土壤中接种病原菌(C+R)的甜菜发病率为80%(附图1A)。
附图1. 本研究实验设计示意图
因为发病率过高导致没有足够的根系材料用于深入的微生物组学分析。因此我们最终只收集了处理:C,S,和S+R的材料用于下一步测序和生物信息学分析见附图2和附表1/2。
附图2. 内生菌群宏基因组分析流程
宏基因组测序结果显示这里有76.1%的序列注释为细菌,10.5%的序列注释为真菌,0.0065%的序列被注释为古细菌(附图3A/B)。
附图3. 质控后数据基因注释的比例
对于真菌序列有监督的cpcoa分析表明内生真菌在三组之间存在显著差异((PERMANOVA), P < 0.05])(附图4A)。这是由于在S+R组大量接种了病原真菌R. solani导致的(附图4B/C)。
附图4. 宏基因组中提取的真菌序列整体分析
A. CSS标准化下的bray-curtis距离;B. 三元图展示真菌读长数量;C. 柱状图展示丝核菌属 Rhizoctonia 的相对丰度
而且其他两组并没有准确注释到真菌的微生物门类。因此,这一结果表明在抑病土壤中接种的R. solani会定殖到甜菜根系,但是却不会发病。
从宏基因组数据中提取16S核糖体保守序列用于注释病评估内生菌细菌群落。结果表明变形菌门和酸杆菌门主导了内生菌细菌群落。这两个菌门中得到的10个OTU均在S+R中显著富集:Pseudomonadaceae (两个), Xanthomonadaceae (四个), Chitinophagaceae (一个), Flavobacteriaceae (两个), 和 Veillonellaceae (一个) (附图5)。
附图5. 甜菜内生菌微生物组的多样性
A. CAP(Constrained Analysis of Principal Coordinates)分析展示Beta多样性(可解释23%的总体差异);conducive soil ©, suppressive soil (S) or suppressive inoculated with R. solani (S+R).
B-D展示门、纲、科水平各组中的相对丰度,其中D图上展示香农多样性指数且无显著差异;
共线性网络分析表明S+R组内生微生物群落复杂度更高(附表3).相关研究也表明连接性比较高的网络往往出现在微生物群落面对逆境的时候,比如病原菌入侵。有意思的是在S+R网络中80%的连接点都属于Chitinophaga, Flavobacterium和 Pseudomonas这三个科。当将属于Bacteroidetes的序列去除,这三组差异就难以区分。这再次暗示了Bacteroidetes 中的 Chitinophaga和Flavobacterium与抑病相关。
图1. 病原菌改变植物内生菌群落结构和功能
Fig. 1. Pathogen-induced changes in endophytic microbiome diversity and functions.
生长在S和S+R两个土壤中的植物内生不同菌群丰度差异
图1A. 通过宏基因组提取16S rRNA基因序列注释细菌群落并统计不同门类细菌差异,最大的圈代表门水平,逐渐缩小的圈按照梯度分别代表纲,科,属。
图1B. 基于宏基因组序列注释功能和物种相关功能差异。最小的圈代表COG功能单位。圆圈大小代表了不同物种或者功能的平均丰度。S组富集的物种或者功能使用绿色标记,S+R组富集的使用蓝色标记。不显著的物种或者功能使用黄色标记。
图1C. 左边散点图描绘了全部S+R组属于拟杆菌门的全部基因/S组对应基因的丰度比值,右边散点图是S组属于拟杆菌门的全部基因/C组对应基因的丰度比值。散点图从上到下排序顺序按照S+R/S比值。每个COG类别缩写对应如下:C:能量代谢;D:细胞周期,细胞分裂和染色体分区;E:氨基酸运输和代谢;F: 核甘酸转运和代谢;G:碳水化合物的运输和代谢;H:辅酶的运输和代谢;I:脂质的运输与代谢;J:翻译,核糖体结构;K:转录;L:复制,重组和修复;M:细胞壁,细胞膜和细胞被膜的生物发生;O:翻译后修饰,蛋白质转换;P:无机离子的运输和代谢;Q:次生代谢产物的生物合成,转运和分解代谢;T:信号转导机制;U:细胞内运输,分泌和囊泡运输;V:防御机制。
内生菌群落功能多样性
Functional diversity of the endophytic microbiome
从宏基因组测序数据中得到50%-70%的序列都被注释到了功能(附图3C-E)。通过对其他基因的注释显示有56,175条与微生物类群相关的功能,其中402个功能是显著在S组中显著富集的。在S+R组我们发现部分基因上调超过10倍,这些基因属于“碳水化合物转运和代谢”和“信号转导机制“。同时发现与这些基因高度相关的微生物门类均在S+R中显著增加,包括: Chitinophagaceae和Flavobacteriaceae (Bacteroidetes); Pseudomonadaceae和 Xanthomonadaceae (Gammaproteobacteria); Hyphomicrobiaceae和Rhizobiaceae (Alpha-proteobacteria); 和* Burkholderiaceae* (Betaproteobacteria) (图1B/C, 和附图9A)。
附图9. 内生菌微生物组碳水化合物活性酶的分布和多样性
A. 热图展示各组中显著差异的基因,按物种来源注释分类
B. 碳水化合物代谢相关的HMM结构域与基因间的相似网络
其中大部分增加的基因(3138/4443) 被注释为与Chitinophagaceae 和Flavobacteriaceae (图1B和附图9A)*相关。当我们选择更严格的阈值定义上调基因时,发现有461个上调基因,这些基因大部分都与*Chitinophagaceae* 和*Flavobacteriaceae**相关。仅对*Bacteroidetes*相关的基因做差异分析,显示了S+R组和S组间按COG功能注释为Q类(次级代谢产物生物合成、转运和分解代谢)的基因差异最大;而S组和C组之间G类(碳水化合物转运和代谢)基因的差异最大(图1C)**。
为了更详细的了解COG的G类和Q类功能的细节,我们分别查找了碳水化合物活性酶数据库(CAZymes)和次级代谢产物生物合成基因簇数据库antiSMASH。通过dbCAN, 我们注释得到1822个基因主要属于一下功能分类模块:糖苷羟化酶、糖基转移酶、多糖裂解酶和糖酯酶结构域以及非催化性糖结合模块。
因为这些结构域与进化相关并且参与相关功能,所以我们使用 hhsearch 算法计算蛋白保守结构域的相似性。从而发现按S+R组内生菌群中有更多的糖苷水解酶和糖基转移酶与抑病相关(图2A和附图9B/C) 。与S组相比,S+R组有三类内生菌在CAZyme酶注释上显著不同 (FDR < 0.1; 图2A和附图9A/B)。 此外,我们发现Chitinophagaceae含有一些与真菌细胞壁降解的相关酶类。比如:几丁质酶、β-葡聚糖酶和内葡聚糖酶等。注释为Burkholderiaceae 和Xanthomonadaceae科(附图9B/C)的菌有两个几丁质酶结构域和三个参与几丁质降解的蛋白酶。这五个结构域在这三种菌是共有的。表明了这些富集的内生菌在同一种功能上的冗余。细菌基因组中包含了大量的BGCs,但是大部分都没有备注到已有的模块和功能。通过antiSMASH分析次级代谢表明730个生物合成基因簇(BGCs)与非核糖体肽、聚酮、萜类、芳基多烯、核糖体合成和翻译后修饰肽(RIPS),膦酸盐、吩嗪和铁载体相关。在这些730个BGCs中已经有12个在先前的研究中报道过,并且物质化学结构也被阐明(附图11和附表5)。
附图11. 内生菌微生物组中已知生物合成基因簇的分布
其中包括两个脂肽类抗生素thanamycin和 brabantamide,并且相关产品也用在土壤中来抑制病原菌Rhizoctonia。其他718个BGCs注释目前还不够成熟,其中117个BGCs显著在S+R组中过表达并且其中34个BGCs属于Bacteroidetes (图3A-F和附图10-12)。值得注意的是这34个BGCs中并不包括先前已经在根际鉴定过的thanamycin和brabantamide。在这过表达的117个基因中有10个NRPS基因簇属于Bacteroidetes菌,并且通过antiSMASH 数据库发现这些都和MIBiG中注释到的基因簇不匹配。
图2. 内生菌群碳水化合物相关活性酶的多样性和分布
Fig. 2. Diversity and distribution of carbohydrate-active enzymes in the endophytic microbiome.
图2A. 与碳水化合物酶类相关的已知蛋白和预测蛋白结构域的相似性网络。使用网络展示不同蛋白结构域之间的距离和相关性。使用CAZymes对生长在抑病土壤S组或接种病原菌的抑病土壤S+R组的植物内生菌群中注释,一共得到1822个基因。节点分为五大类:糖苷水解酶(GH,蓝色);糖基转移酶(GT,橙色);多糖裂解酶(PL,紫色);糖脂酶(CE,绿色)和非催化碳水化合物结合模块(CBM,红色)。位置结构域或者功能尚未验证的蛋白结构域标记为黄色。方形节点代表物种注释为Chitinophagaceae的相关酶在S+R组中显著高于S组。在S+R中过表达的并在分类学水平上属于Burkholderiaceae和Xanthomonadaceae的相关酶类展示在附图9 B/C**中。
图2B. 展示标记的三种富集在S+R组的科水平细菌的CAZymes注释数量。韦恩图黄色代表Burkholderiaceae (黄);蓝色代表 Chitinophagaceae (蓝);绿色代表Xanthomonadaceae (绿)。韦恩图标记了每个酶对应Pfam数据库ID。韦恩图展示和每个物种共有的和特有的相关结构域数量。
从头组装内生菌基因组
De novo assembly of endophytic bacterial genomes
从antiSMASH数据库中鉴定得到的730个BGCs中,有157个包含在组装成的25个基因组 (MAGs)中(附图13/14和附表6)。
附图13. 内生菌微生物组宏基因组数据分箱
A. 重叠群GC含量与覆盖度的散点图;B. 不同分箱软件结果进行ChechM评估完整 性和污染率;C. 重叠群和分箱的长度累计;D. 重叠群的大小和覆盖度样式
附图14. 内生菌的宏基因组分箱与参考基因组比较
A. PCoA展示MAGs和参考基因组中的功能距离;B/C分别代表变形菌门和拟杆菌门类似的分析。
随后根据这些BGCs,我们设计相应的特异性引物来匹配我们分离出来的纯菌。先前我们一共分离得到935个菌株,并使用16S rRNA基因测序鉴定(附图15A/B, 和附表7)。
附图15. 分离内生菌株的物种多样性
A. 属水平内生菌物种组成。B. 培养的937个菌株;C. 拟杆菌门7个测序的基因组、与121已知菌和25个分箱的进化关系。
结果一共有八个不同的属,大部分菌株属于 Bacteroidetes和Gammaproteobacteria。尽管通过PCR,并没有发现BGCs和属于Chitinophaga和 Pseudomonas的菌株之间的联系,但是发现有四个BGCs(BGC298, BGC396, BGC471, and BGC592) 在S+R组富集的内生菌Flavobacterium中。这四个BGCs中有三个 (BGC396, BGC471和 BGC592)属于NRPS基因簇,第四个属于PKS基因簇(BGC298, 图4A)。类似的方法也发现S+R组中获得的三株Chitinophaga菌含有glycosyl hydrolase (GH18)基因。随后这三个菌株用试验验证了具有降解几丁质的能力。对三株Chitinophaga和四株Flavobacterium(同一个属内具有99%相似度)进行基因组测序 (附图15C和16,附表9)。
附图16. 比较基因组分析测序和宏基因组拼接的Flavobacterium基因组
A. Progressive Mauve对齐Flavobacterium基因组。B. 在四2上基因组中生物合成基因簇的结果;C. 基因预测的功能。
结果表明了这些菌的富集的确造成了相关基因的过表达。对于关键的BGCs,与完整的Bacteroidetes基因组比对没有发现我们分箱的基因组是错配造成的(附图16B/C, 附图17A-C)。
附图17. 包含NRPS的生物合成基因簇与MiBIG数据为中已经基因簇的BiG-SCAPE网络
A/B/C分别对应0.3/0.5/0.7一致率的阈值。
图3. 内生菌群落生物合成基因簇的多样性和分布
Fig. 3 Diversity and distribution of biosynthetic gene clusters in the endophytic microbiome.
图3A. 内生菌群中检测到的不同BGCs序列相似性网络(使用BiG-SCAPE构建;阈值选择0.8)。展示节点的物种分类注释和BGC分类注释。在网络中去除少于三条连接的节点(原始网络包含全部的节点 附图10)。节点颜色代表基于Welch’s t test检验的显著性(FDR < 0.1):黄色节点代表不显著;蓝色代表在S+R中显著上调的基因。
图3B. 通过antiSMASH数据库使用Clusterfinder 算法检测到不同处理植物内生菌群过表达的BGCs。
图3C-E. 分别展示变形菌门Proteobacteria ©, 拟杆菌门Bacteroidetes (D) 和末分类unclassified(E)中显著在S+R中富集各类型BGCs的数量(注意:这些BGCs中不包含无法分类的基因)。
图3F. 33个NRPS基因簇的相对丰度聚类热图[使用CSS标准化方法转化RPKM],与C组相比,这些基因簇在S或S+R组中一致表达。
抑病微生物组的重组和功能分析
Reconstruction and functional analysis of disease-suppressive consortia
我们选择了7株菌进行根际定植并测定特定BGCs的表达量。所有的菌株均定植甜菜幼苗的根际和根内。转录组分析也表明了接种病原菌后根际和内生菌群几丁质酶表达量显著增加(图4B/C, 和附图20)。对于这四个基因簇,在植物受到病原菌侵染后,BGC298在根内的表达量比根际更高(*图4C*)。BGC298在四个 *Flavobacterium* 基因组中都存在并且在MIBiG数据库中没有注释(附图17)。
综合以上多个证据表明Flavobacterium 和Chitinophaga在抑病过程中起着重要作用。通过实验验证这一假设,三个独立的验证实验表明了Flavobacterium 和Chitinophaga的组合菌群抑病性能最好。为了证实黄杆菌BGC298在抑病过程中的作用,我们开发了一个SpyCas9介导的基因敲除掉系统。我们获得了两个BGC298的突变体,PKS在这两个突变株中缺失,通过单细胞测序进行了验证。通过这两个突变株同野生株进行定殖实验发现抑病性能降低。并且通过突变株重组菌群的抑病效果也降低。
图4. 抑病微生物群转录和功能分析
Fig. 4. Transcriptional and functional analyses of disease-suppressive consortia.
图4A. 在黄杆菌Flavobacterium MAG nbed44b64 和四株分离的黄杆菌endophytic Flavobacterium中均鉴定到了这四个基因簇:BGC298, BGC396, BGC471和BGC592。图中展示的是NRPS和PKS基因家族编码蛋白的模块类别和保守结构域。结构域和标示如下:C,凝结;A,腺苷酸化;KS,酮合酶;AT,酰基转移酶;PCP,肽载体蛋白;TE,硫酯酶。在GC298中NRPS和PKS基因家族预测底物为甘氨酸、丙二酰辅酶A和甘氨酸。
图4B/C. 通过 Chitinophaga和Flavobacterium 菌株处理甜菜幼苗,使用荧光定量PCR对根际和根内的BGC298, BGC396, BGC471, BGC592和chitinase(GH18) 基因进行表达量的分析。使用 LogRQ进行表达量的展示:低于0表示使用管家基因(glyA)标准化之后下调。柱状图每个柱子为每组重复的均值,黑色线段代表均值的标准差。柱状图上的不同字母代表单因素方差分析和Tukey多重比较后显著的差异。
图4D-F. 使用三株Chitinophaga菌(Ch93, Ch94和 Ch95)和四株Flavobacterium(Fl96, Fl97, Fl98和 Fl5B)单独或者组合验证甜菜发病率,实验表明这些处理菌降低了甜菜枯萎病发病率。
图4D/E. 使用这单独七株菌和七株组合验证发病率。
图4F. 展示敲除了BGC298基因的两种Fl98 突变株 (Fl98-1和Fl98-2);Ch94和Fl98组合菌群和全部七株组合的发病率。
所添加单菌浓度和混合菌株浓度均为107 每克土壤。柱状图每条柱子都是均值,上面展示了标准差的误差线。发病率接种病原菌在21-28天后统计。不同字母表示ANOVA和Tukey HSD检验 多重比较后显著的处理。