本文参考自https://www.jianshu.com/p/25e60508a08f, 原作者为:生信小白2018
基本流程
单拷贝同源基因寻找 → 单基因多序列比对 → 串联后构建发育树
所需软件
- Orthofinder: 寻找单拷贝基因
- MAFFT: 多序列比对
- TRIMAL: 多序列比对修剪
- TRIMAL or FastTree: 串联法(concatenation)构建蛋白质系统发育树
- ASTRAL: 联合/合并法(coalescence)构建系统发育
流程解析
通过Orthofinder寻找单拷贝同源基因
以下内容参考自改文章:「基因组学」使用OrthoFinder进行直系同源基因分析
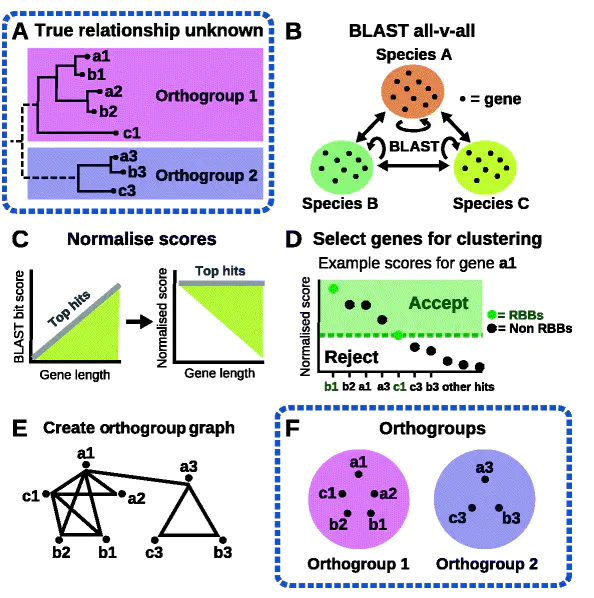
OrthoFinder的分析过程分为如下几步:
- BLAST all-vs-all搜索。使用BLASTP以evalue=10e-3进行搜索,寻找潜在的同源基因。(除了BLAST, 还可以选择DIAMOND和MMSeq2)
- 基于基因长度和系统发育距离对BLAST bit得分进行标准化。
- 使用RBNHs确定同源组序列性相似度的阈值
- 构建直系同源组图(orthogroup graph),用作MCL的输入
- 使用MCL对基因进行聚类,划分直系同源组
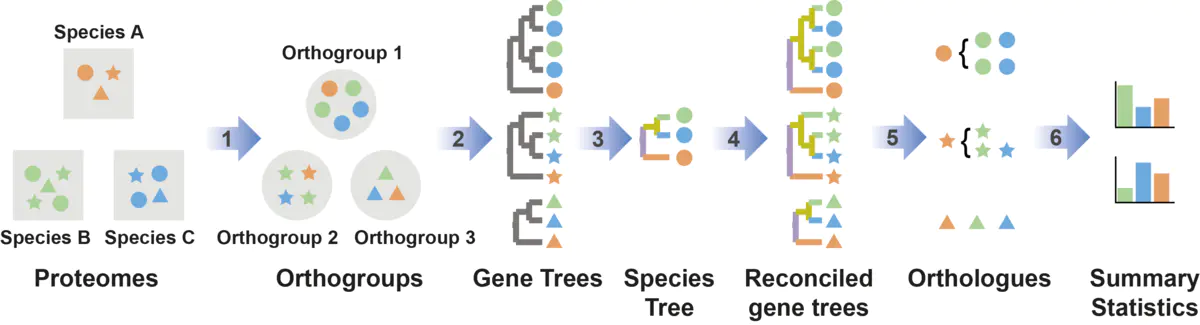
OrthoFinder2在OrthoFinder的基础上增加了物种系统发育树的构建,流程如下:
- 为每个直系同源组构建基因系统发育树
- 使用STAG算法从无根基因树上构建无根物种树
- 使用STRIDE算法构建有根物种树
- 有根物种树进一步辅助构建有根基因树
基于Duplication-Loss-Coalescent 模型,有根基因树可以用来推断物种形成和基因复制事件,最后记录在统计信息中。
OrthoFinder的使用非常方便,一行命令即可,但跑起来比较花时间:
orthofinder -f <folder contains target genomes> -S diamond
OrthoFinder结果:
运行结束后,会在ExampleData里多出一个文件夹,Results_Feb14, 其中Feb14是我运行的日期
直系同源组相关结果文件,将不同的直系同源基因进行分组。
Orthogroups.csv:用制表符分隔的文件,每一行是直系同源基因组对应的基因。
Orthogroups.txt: 类似于Orthogroups.csv,只不过是OrhtoMCL的输出格式
Orthogroups_UnassignedGenes.csv: 格式同Orthogroups.csv,只不过是物种特异性的基因
Orthogroups.GeneCount.csv:格式同Orthogroups.csv, 只不过不再是基因名信息,而是以基因数。
直系同源相关文件,分析每个直系同源基因组里的直系同源基因之间关系,结果会在Orthologues_Feb14文件夹下,其中Feb14是日期
Gene_Trees: 每个直系同源基因基因组里的基因树
Recon_Gene_Trees:使用OrthoFinder duplication-loss coalescent 模型进行发育树推断
Potential_Rooted_Species_Trees: 可能的有根物种树
SpeciesTree_rooted.txt: 从所有包含STAG支持的直系同源组推断的STAG物种树
SpeciesTree_rooted_node_labels.txt: 同上,只不过多了一个标签信息,用于解释基因重复数据。
比较基因组学的相关结果文件:
Orthogroups_SpeciesOverlaps.csv: 不同物种间的同源基因的交集
SingleCopyOrthogroups.txt: 单基因拷贝组的编号
Statistics_Overall.csv:总体统计信息
Statistics_PerSpecies.csv:分物种统计信息
STAG是一种从所有基因推测物种树的算法,不同于使用单拷贝的直系同源基因进行进化树构建。
一些重要概念:
-
Species-specific orthogroup: 一个仅来源于一个物种的直系同源组
-
Single-copy orthogroup: 在直系同源组中,每个物种里面只有一个基因。我们会用单拷贝直系同源组里的基因推断物种树以及其他数据分析。
-
Unassigned gene: 无法和其他基因进行聚类的基因。
-
G50和O50,指的是当你直系同源组按照基因数从大到小进行排列,然后累加,当加入某个组后,累计基因数大于50%的总基因数,那么所需要的直系同源组的数目就是O50,该组的基因树就是G50.
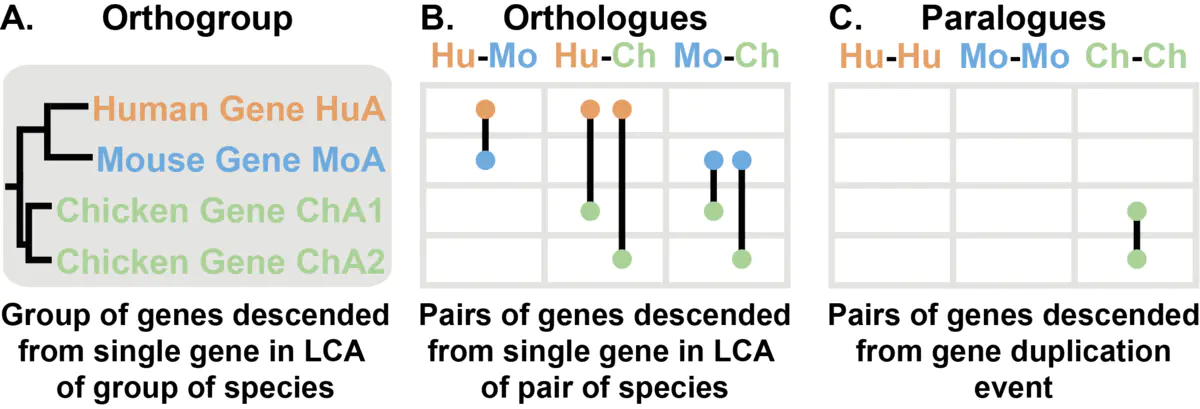
Orthogroups, Orthologs 和 Paralogs 这三个概念推荐看图理解。
- 利用MAFFT进行单同源基因的多序列比对
mafft<input.faa> > <outpuit_aligned.mafft>
- TRIMAL修剪多序列比对结果
trimal -in <input_aligned.mafft> -out <input_aligned.mafft.trimed> -automated1
- Concatenation法进行系统发育分析
将上述trim好的多序列比对结果按照物种顺序进行串联,然后用RaxML或者FastTree进行分析。
raxml -T <thread using> -f a -N <boostrap such as 100> -m <model such as JTT> -x 123456 -p 123456 -s <concatenated_alignment> -n <output.nwk>
FastTree <concatenated_alignment> > <ouput.nwk>
- Coalescence法进行系统发育分析
先用RaxML或者FastTree对每个单拷贝基因进行分析,然后用ASTRAL聚合:
# single gene tree
raxml -T <thread using> -f a -N <boostrap such as 100> -m <model such as JTT> -x 123456 -p 123456 -s <SingleGene_alignment> -n <SingleGene_output.nwk>
# concatenation
cat <all SingleGene_alignment> >> <allSingleGenes_tree.nwk -b>
# bootstrap ana
cat <all bootstrap file> >> <allSingleGenes_bootstrap.txt>
# ASTRAL
java -jar ASTRAL -i allSingleGenes_tree.nwk -b allSingleGenes_bootstrap.txt -r <boostrap> -o <ASTRAL_out.res> >
tail -n 1 <ASTRAL_out.res> > <ASTRAL_out.res.nwk>
集成脚本
该脚本参考自https://github.com/dongwei1220/EasySpeciesTree, 但做了一些适当的修改